For the E.U., Brexit deal is a pretty good one
The Brexit pact preserves crucial E.U. principles like the single market, and lets the bloc look ahead to its future without Britain. — NYT: Top Stories

The Brexit pact preserves crucial E.U. principles like the single market, and lets the bloc look ahead to its future without Britain. — NYT: Top Stories

FILE – In this Aug. 12, 2018 file photo released by the Union for the Republic and Democracy party shows then opposition Presidential candidate Soumaila Cisse casting his ballot during the presidential second round election in Niafunke, Mali. Malian opposition leader Soumaila
2020 has been a year of isolation and grief, but there’s always room for love. — NYT: Top Stories

File-This Jan. 28, 2020, file photo shows a remembrance board at a memorial for Kobe Bryant near Staples Center in Los Angeles. Bryant, the 18-time NBA All-Star who won five championships and became one of the greatest basketball players of his generation
Getting to play Cheetah was even better for the “Saturday Night Live” star, who loves superhero movies: “It was huge on my list of things I wanted to do.” — NYT: Top Stories

Ok, here’s a look at the best holiday-themed television episodes. — FOX News

A mother and child look at the line of trucks parked up on the M20, part of Operation Stack in Ashford, Kent, England, Friday, Dec. 25, 2020. Thousands wait to resume their journey across The Channel after the borders with France reopened.
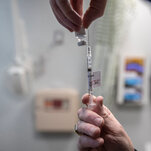
“I am so disappointed and saddened that this happened,” a New York hospital executive wrote to his staff after workers who did not have priority cut the line for the vaccine. — NYT: Top Stories

On a holiday when health experts admonished people not to travel, the weather brought its own hazards across much of the country. — NYT: Top Stories